Weiter geht es mit einem kleinen Fehlschlag:
30 g L-Tryptophan in 240 ml Terpentin bis zum klar werden der Lösung im Rückfluß erhitzt. Nach erkalten braun-gelber Bodensatz im Kolben. LM abdekantiert und Rückstand mit 50 ml Leichtbenzin versetzt. Zugabe von 250 ml 5% AcH unter erwärmen und rühren bis sich Rückstand gelöst hat. Trennen der Phasen im Scheidetrichter, wobei die obere Phase nur leicht gelblich gefärbt war und sich eine gräuliche Interphase ausgebildet hat (diese wurde auch abgetrennt).
Filtrieren und fällen des Tryptamins wie folgt: Zugabe von Natriumhydrogencarbonat, versetzen der Lösung mit 50% NaOH-Lösung und abnutschen des Niederschlags. Beim Trocknen wurde der Rückstand klebrig ölig und roch stark nach Krauseminzenöl.
Rückstand wurde unter erwärmen in Aceton gelöst, wobei sich am Anfang eine klare Lösung bildete, die später ausfiel (feiner Ndg.). Zugabe von 10% Schwefelsäure bis eine klare braune Lösung erhalten wurde, deren Volumen reduziert wurde bis auf eine ölige Flüssigkeit. Beim Eintropfen dieser öligen Lösung in Aceton entstand kein feiner Niederschlag, sondern Klumpen. Durch Zugabe von etwas Wasser zu der öligen Lösung wurde ein feiner weißer Niederschlag erhalten der wiederum abgenutscht wurde. Der Niederschlag löste sich unter gelindem erwärmen in 50 ml Wasser und wurde durch Zugabe von 50% Natronlauge gefällt. Waschen des Ndg. mit Ammoniakwasser und abnutscht.
Niederschlag wird beim Trocknen klebrig Ausbeute: 2.88 g was lediglich 9.6 % entspricht. Zwei DC gefahren:
DC A:
1: 1µl Aceton-Filtrat
2: 1µl L-Tryptophan
3: 0.5µl öliges Tryptamin
4: 1µl Tryptamin p.a.
DC B:
1 und 2: Aceton-Filtrat
3: L-Tryptophan
4: öliges Tryptamin
5 und 6: Eigensynthese
7: Tryptamin p.a.
Laufmittel: 45 ml 94% EtOH plus 5 ml 25% NH3 Lösung
Detektion: 1 Part 10% (m/Vol) DMAB in 25% HCl plus 4 Parts 100% Aceton
Plate: Alugram Sil G/UV254
Aufnahme der Proben in 100% MeOH

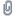 Aceton-Filtrat.jpg (37.05 KB . 593x608 - angeschaut 1814 Mal)
Aceton-Filtrat.jpg (37.05 KB . 593x608 - angeschaut 1814 Mal)Beim DC "B" wurde drauf geachtet die Auftragungspunkte möglichst klein zu bekommen.
Interessanterweise kann eine Umkehrung der Lauflängen beobachtet werden: Tryptamin läuft kürzer als L-Tryptophan. Daher wurde im nächsten DC zum einen das Laufmittel neu angesetzt und zum anderen an Position Nr. 6 die Mixtur #4 hinzugefügt die in 7.5 ml 100% MeOH und 3.5 ml 25% AcH aufgenommen wurde:
1: 1.5µl Aceton-Filtrat (Bottle)
2: 1µl L-Tryptophan (L)
3: 1µl öliges Tryptamin (K)
4: 1µl Eigensynthese (ES)
5: 1µl Tryptamin p.a. (P.A.)
6: 0.5µl Mixture (4)
Neuansatz: Laufmittel: 45ml 94% Brennspiritus + 5 ml 25% NH3-Lösung:
 Aceton-Filtrat2.jpg (15.92 KB . 587x329 - angeschaut 1617 Mal)
Aceton-Filtrat2.jpg (15.92 KB . 587x329 - angeschaut 1617 Mal)Resultat: Der Ammoniak-Gehalt hat Einfluß auf das Laufverhalten von Tryptamin vs. L-Tryptophan. Klingt zwar banal, nur gerät man erst mal ins stutzen, wenn einem sowas passiert.
Mit dem neuen Laufmittel wurde dann ein neues DC gefahren, wo verschiedene Mengen des Aceton-Filtrats aufgetragen wurden:
1: 0.25µl Aceton-Filtrat
2: 0.5µl Aceton-Filtrat
3: 1.5µl Aceton-Filtrat
4: 3µl Aceton-Filtrat
5: 0.5µl L-Tryptophan
6: 0.5µl Eigensynthese
Anmerkung: Die weiße Editierung war ein schön gescannter Teil eines Fingerabdrucks von mir und die Kreise bezeichnen die Blots wo ich eine Fluoreszenzlöschung im UV bekommen habe.
 Aceton-Filtrat3.jpg (19.22 KB . 593x336 - angeschaut 1626 Mal)
Aceton-Filtrat3.jpg (19.22 KB . 593x336 - angeschaut 1626 Mal)Der "abgeschnittene" Punkt vom L-Tryptophan war vermutlich eine Gradientenänderung, da ich in die Kammer kein Filterpapier gelegt habe (keine gesättigte Kammer), zumindest sah man auf dem UV
254nm Bild das da eine zweite Lösemittelfront durchgewandert ist.
Aus den DC´s würde ich sagen, daß mein verschwundenes Tryptamin sich im Aceton-Filtrat wieder gefunden hat, wobei der obere Teil (z.B. bei 0.25µl Aceton-Filtrat) das Tryptamin darstellen dürfte. Stellt sich dann die Frage aus was der untere Blot besteht, denn nicht umgesetztes L-Tryptophan ist in Natronlauge löslich und sollte daher nicht ausgefallen sein.
Lösungsideen:
1. Klebriges Tryptamin: In Chloroform auflösen und nochmals mit Essigsäure extrahieren und mit NaOH fällen, wenn "schönes Pulver" dann ist Chloroform zur Reinigung unbedingt nötig.
2. Sulfat vs. Hydrochlorid: Je 1 g Tryptamin (Eigensynthese) mit 24% Salzsäure und 50% Schwefelsäure umsetzen, beide Lösungen dann mit Aceton fällen, Niederschlag trocken und auswiegen, anschließend auf Tryptamin zurückrechnen und schauen wie die Ausbeuten sind. Zusätzlich DC´s von den Aceton-Filtraten.
3. Aceton-Filtrat aus dieser Synthese: Aceton im Roti abziehen und Rückstand in Chloroform aufnehmen. Danach je nach Verhalten der Chloroform-Phase Weiterverarbeitung: Klare Lösung=Ausschüttlen mit Wasser und fällen mit NaOH oder bei Niederschlag: Abfiltrieren desselben und verwerfen des Chloroforms, Ndg. in Wasser lösen und fällen mit NaOH.
bombjack
PS: Meine DC-Platten sind alle und die Packungen die ich noch habe sind mindestens 10 Jahre alt...d.h. neue Validierung ist angesagt....

